Presentation link
My research at PLU
Funding supported research:
-
J-term 2019 - Spring 2019
[Collaborative Project]AI and Smart Home Technologies for Promoting Energy Efficiency and Conservation in the Pacific Northwest
TBD participants
My main research at PLU is mainly focus on Capstone projects with undergraduate students.
-
Fall 2020 - Spring 2021
[Capstone Project]:Stock Prediction Tool Based on Machine Learning and Natural Language Processing
Marcus Lee, Erika Powell, Vanessa Hoang, Nolan Premo
-
Fall 2020 - Spring 2021
[Capstone Project]:Creation of Bioinformatics Tools Web Server using Cloud Computing
Kyle Hippe, Christian Oakley, Daniel Shin
-
Fall 2019 - Spring 2020
[Capstone Project]:Studyhub - A Study Buddy Networking Application
Chris Caudill, Holden Gjuka
-
Fall 2018 - Spring 2019
[Capstone Project]:Automated Modeling of HIV Treatment Data
Emily Shane, John Smith, Natalie Stephenson
-
Fall 2018 - Spring 2019
[Capstone Project]:Poisonous Plant Recognition
Dawson Faker , Jacob Leigh, Caroline Powell
-
Summer 2018 -
[Summer Research Project]:Machine learning on protein model quality assessment
Max staples, Natalie Stephenson
-
Fall 2017 - Spring 2018
[Capstone Project]:AI on Kinect
Paul Jett (BS), David Stoppenbrink (BS), John Woelfel (BS)
-
Fall 2017 - Spring 2018
[Capstone Project]:AI on Geoscience, co-advised by Prof. Wilcox
Zach Golden (BS), Boen Zhang (BS)
-
Fall 2017 - Spring 2018
[Capstone Project]:AI on Plant
"Kyle Bendebel (BS), Joel Goh (BA), David Ries (BS)"
-
Summer 2017 -
[Summer Research Project]:Machine learning on protein model quality assessment
Matthew Conover, John Smith
-
Fall 2016 - Spring 2017
[Capstone Project]:Computational methods for protein function prediction on CAFA
Caleb Chandler, Miguel Amezola, Devon Johnson
-
Spring 2017 -
[Research Project]:Computational methods for protein function prediction on CAFA
Colton Freitas
-
Spring 2017 -
[Research Project]:Application of machine learning methods
Nathan Kosylo, Joshua Moran, Yiming Gan
My research at MU
My research is mainly focus on applying machine learning and data mining techniques to address the biomedical problems, such as gene function prediction from Hi-C contact data, protein function prediction, quality assessment for protein tertiary structure prediction, and etc.
Gene function prediction
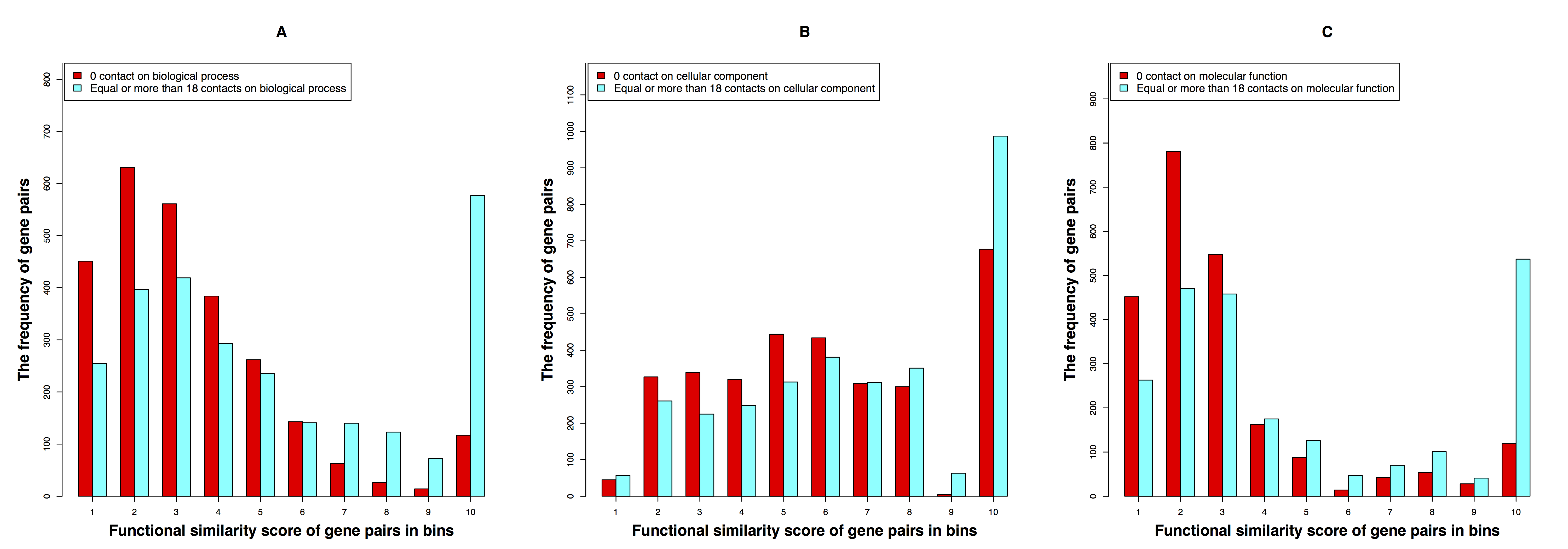
Hi-C technique (Z. Wang, R. Cao, et. al, 2013) that can determine the genome-wide chromosomal interaction/contact data was used for spatial proximity profiles of healthy and malignant human B cell/cell-lines. We use it for generating the spatial gene-gene interaction networks, and compare the function similarity of gene pairs that do not spatially interact and that have interactions. We find out that genes having strong spatial interactions tend to have highly similar function in terms of biological process, molecular function and cellular component of the Gene Ontology. And even though the level of gene-gene interactions generally have no or weak correlation with either sequential genomic distance or sequence identity between genes, the interacted genes with high function similarity tend to have stronger interactions, somewhat shorter genomic distance and significantly higher sequence identity. We develop and evaluate a new gene function prediction method based on gene-gene interacting networks, which can predict gene function well for a large number of human genes. Details can be found at this reference (R. Cao et. al. 2015.).
Protein function prediction
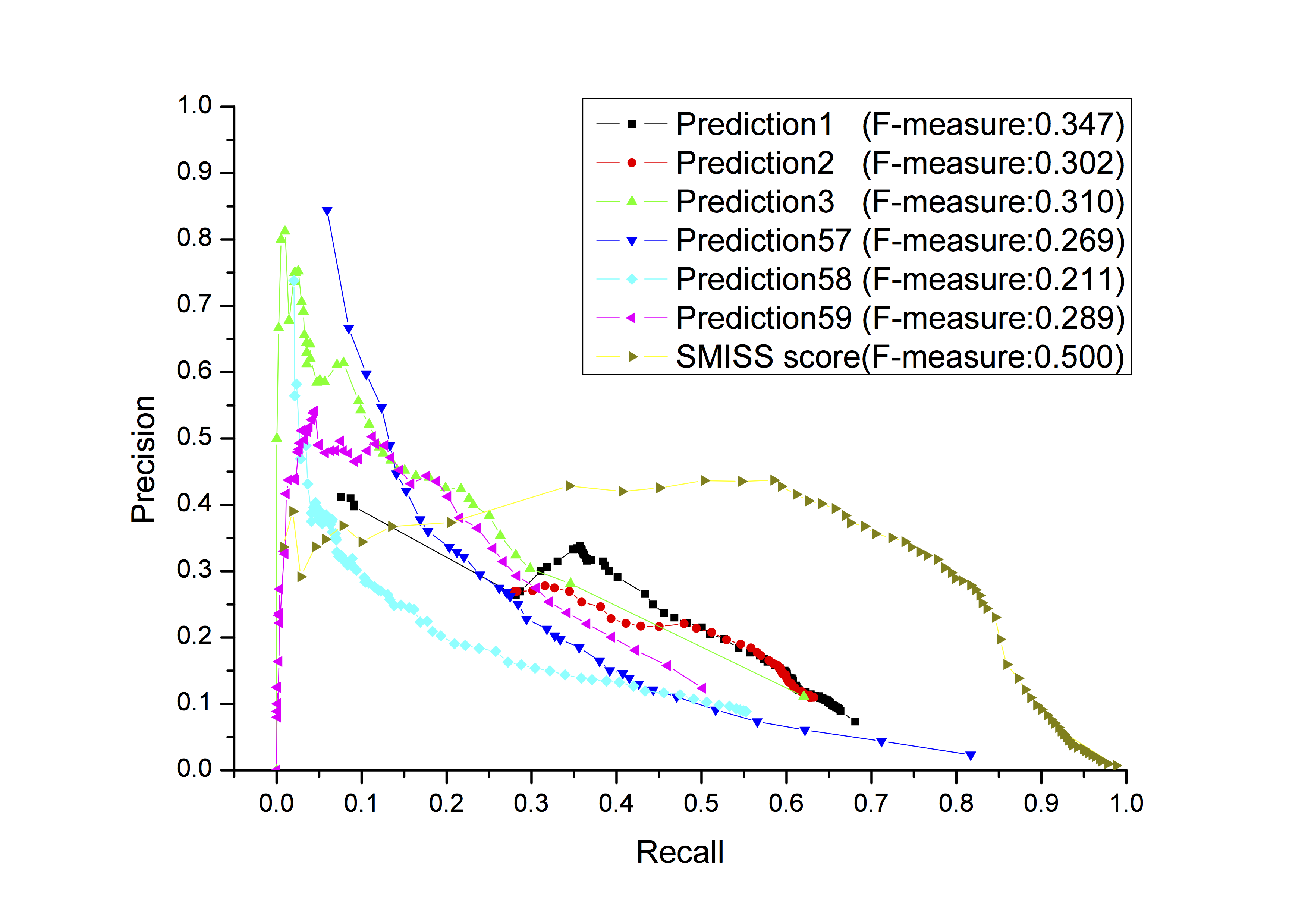
Functionally relevant biological information such as protein sequences, gene expression, and protein-protein interactions has been used mostly separately for protein function prediction. One of the major challenges is how to effectively integrate multiple sources of both traditional and new information such as spatial gene-gene interaction networks generated from chromosomal conformation data together to improve protein function prediction. We developed three different probabilistic scores (MIS, SEQ, and NET score) from protein sequence, function associations, and protein-protein interaction and spatial gene-gene interaction networks for protein function prediction. These three scores were combined in a new Statistical Multiple Integrative Scoring System (SMISS) to predict protein function. More details at the reference (R. Cao, et. al. 2015.).
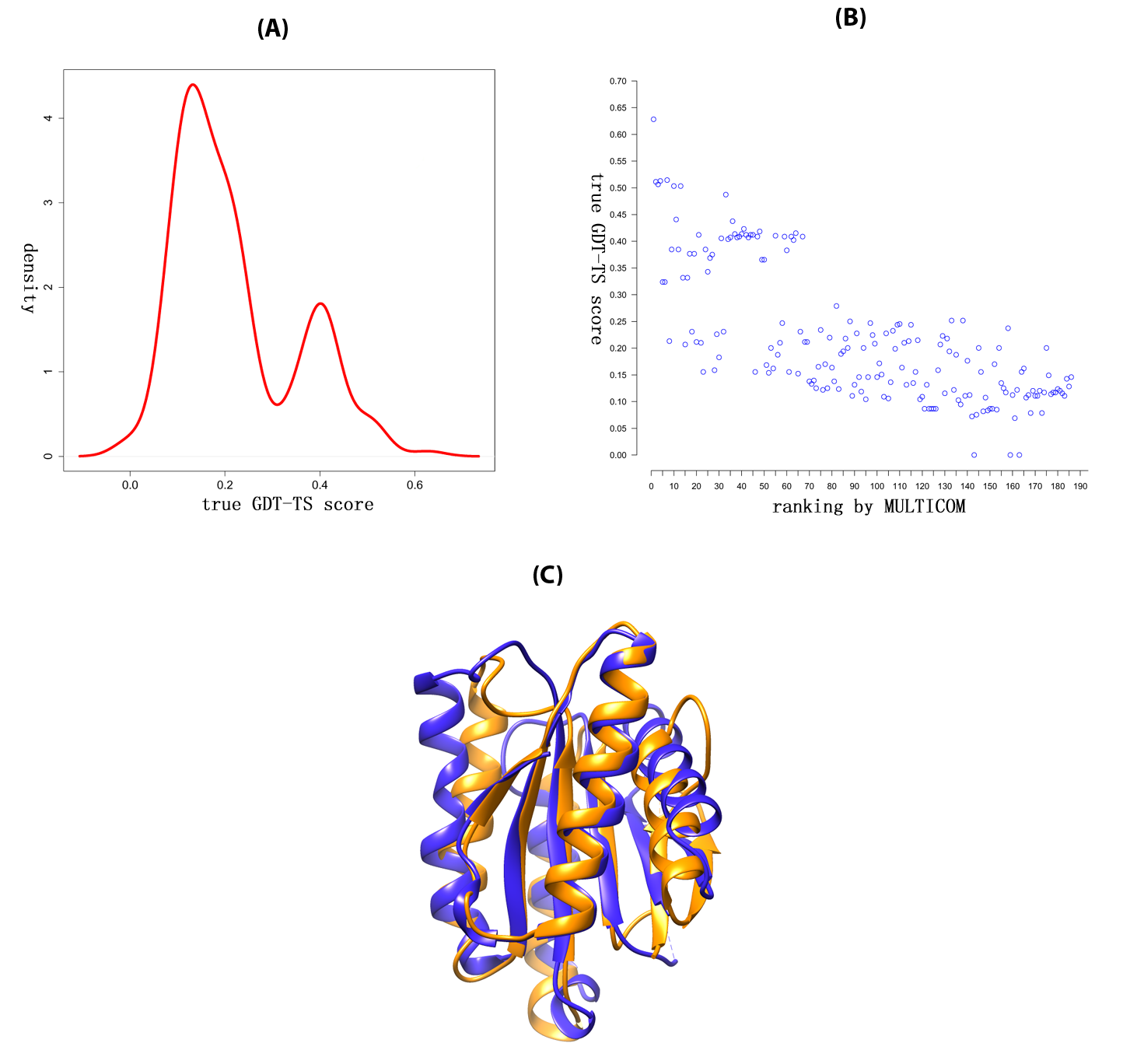
Quality assessment for protein structure prediction
Quality assessment is to evaluate the quality of a protein model without knowing the native structure, which is crucial for protein tertiary structure prediction. We first evaluate our MULTICOM QA methods on CASP10, and the results show that the pairwise model assessment methods worked better when a large portion of models in the pool were of good quality, whereas single-model quality assessment methods performed better on some hard targets when only a small portion of models in the pool were of reasonable quality. (See R. Cao, et. al. 2014.) And then we developed a machine learning QA tool SMOQ (See R. Cao, et. al. 2014.), which can predict the distance deviation of each residue in a single protein model. In addition, we developed a novel large-scale model QA method in conjunction with model clustering to rank and select protein structural models. It unprecedentedly applied 14 model QA methods to generate consensus model rankings, followed by model refinement based on model combination (i.e. averaging). Our experiment demonstrates that the large-scale model QA approach is more consistent and robust in selecting models of better quality than any individual QA method. It was offically ranked 3rd out of all 143 human and server predictors in CASP11 (See R. Cao, et. al. 2015., R. Cao, et. al. 2015.).